FDA genehmigt randomisierte kontrollierte RECOVER IV-Studie mit Ausnahme der Einwilligungserklärung (EFIC)
FDA genehmigt randomisierte kontrollierte RECOVER IV-Studie mit Ausnahme der Einwilligungserklärung (EFIC)
FDA genehmigt auch RECOVER III Post-Approval-Studie und schließt sie ab
DANVERS, Massachusetts, USA--(BUSINESS WIRE)--Abiomed (Nasdaq: ABMD) gibt bekannt, dass die US-Arzneimittelbehörde FDA zwei Zulassungen für die klinische Forschung mit Impella-Herzpumpen bei Patienten mit kardiogenem Schock nach akutem Myokardinfarkt (AMI) erteilt hat.
Die FDA hat die randomisierte kontrollierte Studie (RCT für Randomized Controlled Trial) (On-Label) RECOVER IV für Patienten mit kardiogenem Schock nach einem AMI genehmigt. RECOVER IV ist eine zweiarmige Studie, in der untersucht werden soll, ob eine perkutane Koronarintervention (PCI) mit Impella-Unterstützung, die vor der PCI eingeleitet wird, einer PCI ohne Impella-Unterstützung überlegen ist.
„Diese bahnbrechende Studie ist der Höhepunkt von mehr als 20 Jahren Forschung in der interventionellen Therapie des AMI und wird alle klinischen Fortschritte anwenden, die wir gemacht haben, um das Überleben und die Erholung des Herzens von Patienten mit kardiogenem Schock nach einem AMI zu verbessern, wie in mehreren prospektiven Studien gezeigt wurde“, sagte William O'Neill, MD, medizinischer Direktor des Zentrums für strukturelle Herzkrankheiten bei Henry Ford Health und einer der Hauptprüfer von RECOVER IV.
Der primäre Endpunkt von RECOVER IV ist die Gesamtmortalität nach 30 Tagen. Zu den sekundären Endpunkten gehören schwerwiegende unerwünschte kardiovaskuläre und zerebrovaskuläre Ereignisse (MACCE) nach 30 Tagen, Überleben außerhalb des Krankenhauses nach sechs Monaten, Wiederherstellung der linksventrikulären (LV-) Funktion, Notwendigkeit eines dauerhaften ventrikulären Unterstützungsgeräts (VAD) oder einer Herztransplantation und gesundheitsbezogene Lebensqualität, gemessen anhand der Antworten auf den Kansas City Cardiomyopathy Questionnaire (KCCQ) nach einem Jahr. Das Ziel von Abiomed bei der Durchführung der Studie ist es, eine globale Richtlinienempfehlung der Klasse I für kardiogenen Schock nach einem AMI für Impella und damit verbundene Best-Practice-Protokolle zu erreichen, einschließlich der Impella-Implantation vor einer PCI (siehe Abbildung 1).
„Ich bin optimistisch, dass RECOVER IV die Vorteile der hämodynamischen Unterstützung und der Best-Practice-Protokolle weiter verdeutlichen wird. Zu diesen Vorteilen gehören die ventrikuläre Entlastung mit Impella vor der PCI, eine verringerte LV-Wandbelastung, eine verringerte pulmonale Stauung, ein verstärkter kollateraler koronarer Blutfluss und ein verbesserter Kardioschutz, so dass mehr Patienten mit kardiogenem Schock nach einem AMI überleben und eine Erholung des nativen Herzens erreichen können. Das Herzteam und die Fachwelt haben sich weiterentwickelt und verstehen, wie wichtig die Erholung des Herzmuskels sowohl bei AMI als auch bei kardiogenem Schock bei AMI ist, um die wachsende Epidemie der Herzinsuffizienz einzudämmen“, sagte Navin K. Kapur, MD, Executive Director des Cardiovascular Center for Research and Innovation (CVCRI) am Tufts Medical Center und nationaler Co-Principal Investigator für RECOVER IV.
FDA genehmigt und schließt Post-Approval-Studie RECOVER III ab
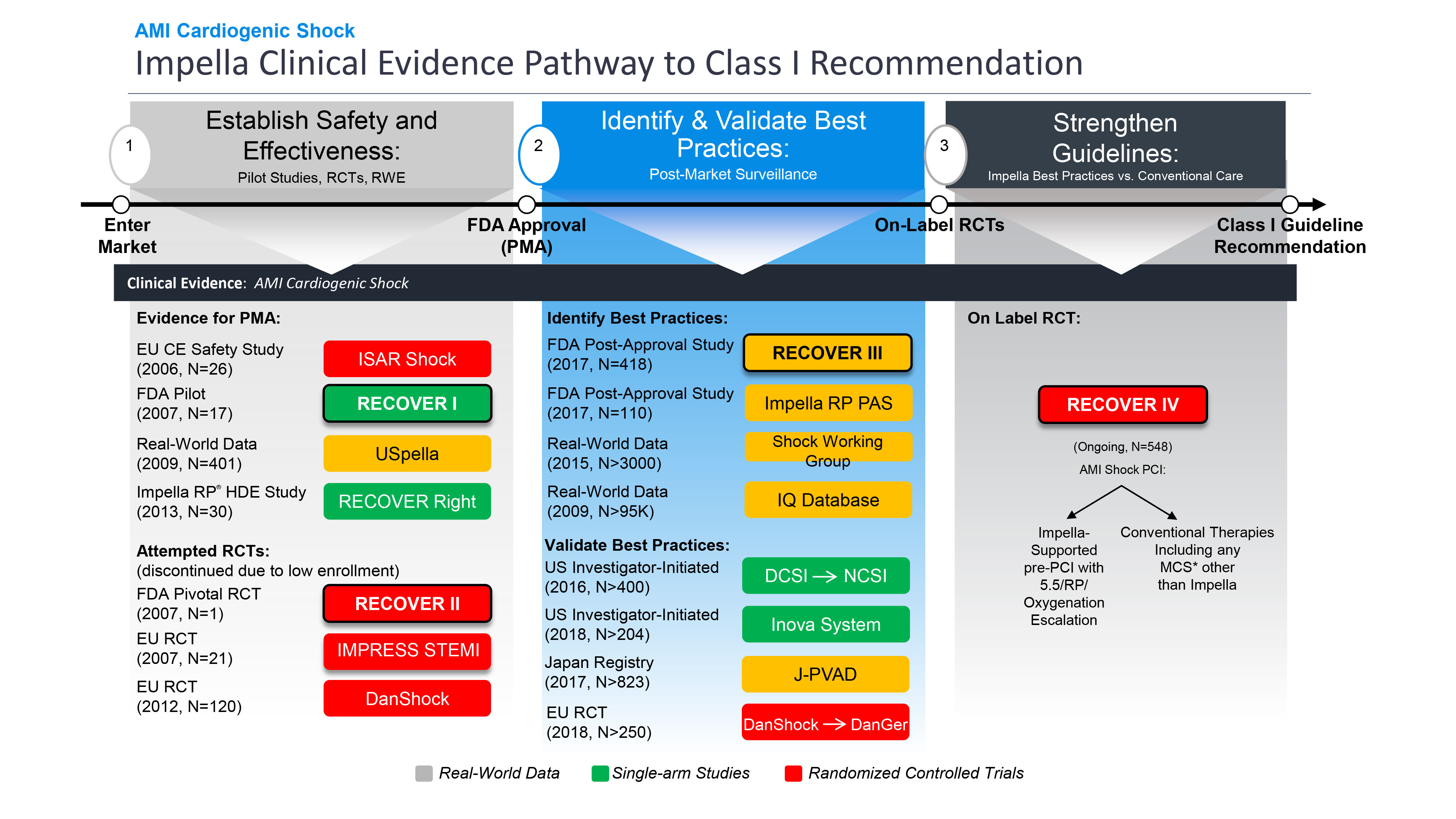
Darüber hinaus hat die FDA die prospektive Post-Approval-Studie (PAS) RECOVER III zu Impella bei kardiogenem Schock nach einem AMI genehmigt und abgeschlossen. Im Rahmen dieser Studie wurden reale Daten zu Patienten mit kardiogenem Schock nach einem AMI gesammelt, die zwischen 2017 und 2019 mit Impella behandelt wurden, und es wurden detaillierte Daten erhoben, darunter die Stadien des kardiogenen Schocks, die Herzleistung und der Zeitpunkt der Implantation. RECOVER III erfüllt die PAS-Anforderung von Abiomed, und die Genehmigung sowie der Abschluss von RECOVER III durch die FDA validiert Impella als sichere und wirksame Therapie für den kardiogenen Schock bei AMI weiter.
Impella ist nach wie vor das einzige Gerät zur mechanischen Kreislaufunterstützung (MCS), das von der FDA die höchste Stufe der Zulassung vor der Markteinführung (PMA) und die PAS-Zulassung für den kardiogenen Schock nach einem AMI erhalten hat. Auf der Grundlage von RECOVER III wird die Kennzeichnung von Impella für den kardiogenen Schock nach einem AMI aktualisiert, um Daten für den Zeitraum von bis zu einem Jahr nach dem Eingriff zu berücksichtigen.
Klinische Anamnese für kardiogenen Schock nach AMI
Der kardiogene Schock bei AMI hat eine der höchsten Sterblichkeitsraten im medizinischen Bereich. Die Überlebensrate von Patienten mit kardiogenem Schock in den SCAI-Stadien D und E liegt ohne Impella-Unterstützung und damit verbundene bewährte Verfahren weiterhin bei etwa 50 %. Das Überleben allein ist bei kardiogenem Schock nicht mehr der Goldstandard. Mehrere Best-Practice-Studien zu Impella zeigen eine Überlebensrate von mehr als 70 % bei einer Erholung des nativen Herzens von mehr als 90 % (siehe Abbildung 2). Die Best Practices für Impella, wie z. B. die Implantation vor der PCI (siehe Abbildung 3), wurden von anerkannten medizinischen Experten auf dem Gebiet der Kreislaufunterstützung entwickelt und in den letzten zehn Jahren in zahlreichen klinischen Studien in den USA, Deutschland, Italien und Japan veröffentlicht. Die Erholung des Herzens nach einem kardiogenen Schock verbessert die Lebensqualität der Patienten und macht Impella zu einer der kosteneffektivsten Therapien in der CMS Medicare Population und in der Privatversicherung. Allein in den USA werden jedes Jahr mehr als 200.000 Patienten mit einem kardiogenen Schock ins Krankenhaus eingeliefert
FDA-Zulassungsgeschichte des kardiogenen Schocks bei AMI
Impella ist die am meisten untersuchte Herzpumpe in der Geschichte der FDA (siehe Abbildung 4 und 5) und hat eine exklusive PMA der FDA als sichere und wirksame Therapie für kardiogenen Schock, Hochrisiko-PCI und Rechtsherzversagen. Seit 2004 wurden mehr als 1.200 von Experten begutachtete Studien über den klinischen Nutzen von Impella veröffentlicht, darunter Analysen der real-world evidence, prospektive klinische Studien und RCTs.
Impella wurde weltweit zur Behandlung von mehr als 235.000 Patienten eingesetzt und ist in 13 Leitlinien klinischer Fachgesellschaften enthalten. Im Jahr 2021 stufte die Europäische Gesellschaft für Kardiologie Impella auf eine Empfehlung der Klasse IIa für die Behandlung des kardiogenen Schocks hoch. Der intraaortale Ballon (IAB) wird derzeit in Europa und Japan für den routinemäßigen Einsatz bei kardiogenem Schock als Klasse III (schädlich) eingestuft, und zwar auf der Grundlage der RCT IABP-SHOCK II, die zeigte, dass der IAB im Vergleich zur inotropen Therapie keinen Nutzen für das Überleben oder die hämodynamische Verbesserung bietet. Im Jahr 2020 wurde der IAB in den US-amerikanischen Leitlinienempfehlungen für den kardiogenen Schock nach einer Kardiotomie in die Klasse III (schädlich) eingestuft.
Alle MCS und ventrikulären Unterstützungssysteme (VADs) seit 1992 wurden mit einarmigen Studien zugelassen, in denen die historischen Überlebensraten anhand objektiver Leistungskriterien (OPC) verglichen wurden, da die Randomisierung kritisch kranker Patienten, die eine sofortige hämodynamische Verstärkung benötigen, eine ethische und logistische Herausforderung darstellt. In den Jahren 2008 und 2009 unternahm Abiomed einen Versuch im Rahmen der FDA-RCT RECOVER II, in der Impella mit IAB bei kardiogenem Schock nach AMI verglichen wurde. In RECOVER II wurde in 15 Monaten an mehr als 30 Standorten nur ein Patient aufgenommen, bevor die Studie wegen logistischer und ethischer Probleme bei der Einwilligung und mangelnder Teilnehmerzahl abgebrochen wurde.
Die FDA erteilte Impella im Jahr 2008 die 510(k)-Zulassung. Nach mehreren von der FDA und von Ärzten initiierten prospektiven Studien erteilte die FDA 2015 die Zulassung für Hochrisiko-PCI, 2016 für kardiogenen Schock bei AMI (siehe Abbildung 6) und 2018 für andere Formen der Herzinsuffizienz mit kardiogenem Schock. Mit dem Abschluss von RECOVER III und der Zulassung von RECOVER IV RCT verfolgt Abiomed eine On-Label-Studie, um die globalen Leitlinien zu stärken und die Ergebnisse für Patienten zu verbessern.
Abiomed hat seit 2006 mehrere Studien zum kardiogenen Schock bei AMI gesponsert und finanziert (siehe Abbildung 7), darunter die einzigen FDA-Studien in diesem Bereich. Die Schwierigkeit, Patienten mit kardiogenem Schock nach einem AMI zu randomisieren, wurde in mehreren Studien nachgewiesen, darunter IMPRESS in STEMI (n=18), IMPRESS in Cardiac Arrest (n=48), Seyfarth et al. (n=26) und die von Abiomed gesponserte FDA RECOVER II RCT (n=1). Alle diese Studien konnten nicht randomisiert werden und wurden vorzeitig abgebrochen, weil die vorgesehene Teilnehmerzahl aufgrund logistischer und ethischer Probleme nicht erreicht wurde.
Neue FDA-Regelungsänderungen für Ausnahmen von der Einwilligungserklärung (EFIC):
Die Randomisierung von AMICS-Patienten (Patienten mit kardiogenem Schock nach einem AMI) stellt eine Herausforderung dar, da sie eine Notfallversorgung benötigen und zu krank sind, um eine traditionelle informierte Zustimmung zur Teilnahme an einer Studie zu geben. 1996 führte die FDA den Weg der Ausnahme von der informierten Zustimmung ((Exception from Informed Consent) für die klinische Notfallforschung ein. Auf diese Weise können Prüfer eine Gemeinschaft umfassend über eine Studie informieren und dann Patienten ohne die Zustimmung der Patienten, ihrer Familie oder ihrer gesetzlichen Vertreter einschreiben.
Im Jahr 2022 genehmigte die FDA - nach Rücksprache mit der FDA über den Studienentwurf der RECOVER IV RCT - das Studienprotokoll der RECOVER IV RCT, das den Einsatz von EFIC beinhaltet. Dieses Verfahren zur Sensibilisierung der Gemeinschaft ist selten und wird nur angewandt, wenn die zu untersuchenden Patienten unter einer lebensbedrohlichen Erkrankung leiden, die zu einer schweren Beeinträchtigung der geistigen Leistungsfähigkeit führt. Dies ist ein wichtiger Meilenstein für das Fachgebiet und die führenden Ärzte. Zu den nächsten Schritten gehören die Genehmigung durch das Institutional Review Board (IRB) des örtlichen Krankenhauses und die Verpflichtung der Ärzte zur Randomisierung.
„Diese entscheidende randomisierte Studie ist von historischer Bedeutung, da sie die erste ist, bei der die EFIC-Gemeinschaftseinwilligung zur Rekrutierung von Patienten mit kardiogenem Schock verwendet wird. Ich freue mich über die Partnerschaft der FDA mit dem Ziel, die Herausforderungen bei der Einwilligung in RCTs zum kardiogenen Schock zu bewältigen, und fordere die Ärzteschaft auf, Patienten in RECOVER IV einzuschreiben und zu randomisieren“, sagte Gregg W. Stone, MD, Professor für Medizin und Direktor für akademische Angelegenheiten des Mount Sinai Heart Health System in New York sowie Studienleiter für RECOVER IV.
Für weitere Informationen über die derzeit besten Praktiken bei der Behandlung von Patienten mit kardiogenem Schock nach einem AMI klicken Sie hier.
Für weitere Informationen über die RECOVER IV RCT-Studie klicken Sie hier.
ÜBER IMPELLA-HERZPUMPEN
Impella 2.5® und Impella CP® sind von der US-amerikanischen FDA für die Behandlung von Patienten mit bestimmten fortgeschrittenen Herzerkrankungen zugelassen, die einer elektiven und dringlichen perkutanen Koronarintervention (PCI: Percutaneous Coronary Intervention) wie z. B. Stentimplantation oder Ballon-Angioplastie unterzogen werden, um verstopfte Koronararterien freizumachen.
Impella 2.5, Impella CP, Impella CP mit SmartAssist®, Impella 5.0®, Impella LD® und Impella 5.5® mit Smart Assist® sind von der FDA zur Behandlung von Herzinfarkt- oder Kardiomyopathie-Patienten mit kardiogenem Schock zugelassen. Diese einzigartigen Herzpumpen ermöglichen eine Wiederherstellung der nativen Herzfunktion, sodass die Patienten mit ihrem eigenen Herzen nach Hause entlassen werden können.
ÜBER ABIOMED
Abiomed, Inc. mit Sitz in Danvers, Massachusetts (USA), ist ein führender Anbieter von Medizintechnik zur Kreislaufunterstützung und Oxygenierung. Unsere Produkte entlasten das Herz durch die Verbesserung des Blutflusses und/oder stellen eine ausreichende Sauerstoffversorgung bei Patienten mit Atemwegsversagen sicher. Weitere Informationen erhalten Sie unter: https://protect-us.mimecast.com/s/sBTlCn58zLHnZBnjS0aCEg?domain=abiomed.com.
ZUKUNFTSGERICHTETE AUSSAGEN
Zukunftsgerichtete Aussagen unterliegen Risiken und Unwägbarkeiten, wie sie in den periodischen Berichten von Abiomed beschrieben werden, die bei der Securities and Exchange Commission hinterlegt sind. Die tatsächlichen Ergebnisse können erheblich von den erwarteten Ergebnissen abweichen.
Die Ausgangssprache, in der der Originaltext veröffentlicht wird, ist die offizielle und autorisierte Version. Übersetzungen werden zur besseren Verständigung mitgeliefert. Nur die Sprachversion, die im Original veröffentlicht wurde, ist rechtsgültig. Gleichen Sie deshalb Übersetzungen mit der originalen Sprachversion der Veröffentlichung ab.
Contacts
Medienkontakt:
Jenny Leary
Associate Director, U.S. Communications
+1 (978) 882-8491
jleary@abiomed.com
Investorenkontakt:
Todd Trapp
Executive Vice President und Chief Financial Officer
+1 (978) 646-1680
ttrapp@abiomed.com