

Newsroom
Sorted by: Latest
-
Alchemer Positioned as a Challenger in the 2026 Gartner® Magic Quadrant™ for Voice of the Customer Platforms
LOUISVILLE, Colo.--(BUSINESS WIRE)--Alchemer today announced that it has been positioned by Gartner as a Challenger in the Magic Quadrant for Voice of the Customer Platforms....
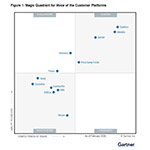
-
Medallia Named a Leader in the 2026 Gartner® Magic Quadrant™ for Voice of the Customer Platforms Report
TYSONS, Va.--(BUSINESS WIRE)--Medallia, Inc., the global leader in customer and employee experience, today announced that it has been named a Leader for the fifth consecutive publication in the Gartner® Magic Quadrant™ for Voice of the Customer (VoC) Platforms report. Medallia was recognized in the report for its Completeness of Vision and Ability to Execute. “We believe Medallia’s recognition as a Leader reflects our mission to turn massive volumes of customer data into meaningful business out...
-
Core Development Group Sponsors Reading Event at Boys & Girls Club of Paterson and Passaic
MAHWAH, N.J.--(BUSINESS WIRE)--Core Development Group, a leading New Jersey-based commercial solar and energy storage contractor, today announced its premium sponsorship of the Boys & Girls Club of Paterson and Passaic’s Read Across America event. Read Across America is an annual national reading motivation and awareness event scheduled to take place March 9–13, aligning with Dr. Seuss’s birthday, and serves as a nationwide, week-long celebration to boost literacy among children. Hosted at...

-
Cornerstone Building Brands Manufacturing Leader Receives 2026 STEP Ahead Award from The Manufacturing Institute
CARY, N.C.--(BUSINESS WIRE)--Cornerstone Building Brands quality and technology manager named a 2026 STEP Ahead Award honoree by The Manufacturing Institute....

-
Xtrackers UK Regulatory Announcement: Net Asset Value(s)
LONDON--(BUSINESS WIRE)-- Xtrackers Investment company with variable capital (Société d'investissement à capital variable) Registered office: 49, avenue J.F. Kennedy, L-1855 Luxembourg R.C.S. Luxembourg B-119.899 (the “Company”) NOTICE TO SHAREHOLDERS OF THE SUB-FUNDS LISTED ON THE LONDON STOCK EXCHANGE Luxembourg, 11 March 2026 The board of directors of the Company (the “Board of Directors”) hereby informs all shareholders of the sub-funds listed in the table below (each a “Sub-Fund”, and tog...
-
CACI Closes $500 Million Offering of 6.375% Senior Notes
RESTON, Va.--(BUSINESS WIRE)--CACI International Inc (NYSE: CACI) today announced it closed a $500 million offering of 6.375% unsecured senior notes due in 2033 (the “notes”). The notes will be issued as part of the same series as CACI’s 6.375% senior notes due 2033 originally issued in June 2025. CACI intends to use the net proceeds from the offering to repay certain indebtedness under its revolving credit facility that was incurred to pay a portion of the purchase price of its acquisition of...

-
Weiss Asset Management LP UK Regulatory Announcement: Form 8.3
BOSTON--(BUSINESS WIRE)-- FORM 8.3 PUBLIC OPENING POSITION DISCLOSURE/DEALING DISCLOSURE BY A PERSON WITH INTERESTS IN RELEVANT SECURITIES REPRESENTING 1% OR MORE Rule 8.3 of the Takeover Code (the "Code") 1. KEY INFORMATION (a) Full name of discloser: Weiss Asset Management LP (Investment Manager of Brookdale International Partners, L.P. and Brookdale Global Opportunity Fund) (b) Owner or controller of interests and short positions disclosed, if different from 1(a): The naming of nominee or v...
-
Millennium Partners, L.P. UK Regulatory Announcement: Form 8.3 - Amendment
LONDON--(BUSINESS WIRE)-- AMENDMENT TO SECTION 2(a) OF FORM 8.3 (SEQUENCE NUMBER 1549119) PUBLISHED AT 13:56 UTC ON MARCH 11TH, 2026 FORM 8.3 PUBLIC OPENING POSITION DISCLOSURE/DEALING DISCLOSURE BY A PERSON WITH INTERESTS IN RELEVANT SECURITIES REPRESENTING 1% OR MORE Rule 8.3 of the Takeover Code (the “Code”) 1. KEY INFORMATION (a) Full name of discloser: Millennium International Management LP (b) Owner or controller of interests and short positions disclosed, if different from 1(a): The n...
-
Millennium Partners, L.P. UK Regulatory Announcement: Form 8.3
LONDON--(BUSINESS WIRE)-- FORM 8.3 PUBLIC OPENING POSITION DISCLOSURE/DEALING DISCLOSURE BY A PERSON WITH INTERESTS IN RELEVANT SECURITIES REPRESENTING 1% OR MORE Rule 8.3 of the Takeover Code (the “Code”) 1. KEY INFORMATION (a) Full name of discloser: Millennium International Management LP (b) Owner or controller of interests and short positions disclosed, if different from 1(a): The naming of nominee or vehicle companies is insufficient. For a trust, the trustee(s), settlor and beneficiari...
-
Millennium Partners, L.P. UK Regulatory Announcement: Form 8.3
LONDON--(BUSINESS WIRE)-- FORM 8.3 PUBLIC OPENING POSITION DISCLOSURE/DEALING DISCLOSURE BY A PERSON WITH INTERESTS IN RELEVANT SECURITIES REPRESENTING 1% OR MORE Rule 8.3 of the Takeover Code (the “Code”) 1. KEY INFORMATION (a) Full name of discloser: Millennium International Management LP (b) Owner or controller of interests and short positions disclosed, if different from 1(a): The naming of nominee or vehicle companies is insufficient. For a trust, the trustee(s), settlor and beneficiari...